Plant Metabolic Networks and their Organization
The first characteristic feature of plant metabolism is its biosynthetic capacity (Croteau et al.,2000; Wink, 1999).While bacterial and yeast metabolisms encompass only a few hundred metabolites, the number of known plant secondary products is estimated to be 100,000 (Schwab, 2003), and the actual number may be as high as 200,000 (Sumner et al., 2003). Obviously individual species synthesize only a particular subset of these compounds, but any attempt to define the metabolic network in a plant cell has to include substantially more biosynthetic pathways than in a typical microorganism. Moreover, since the manipulation of the fluxes through these pathways can be of agronomic and commercial interest (Dixon and Sumner, 2003), the definition of the secondary pathways in the metabolic network may be as important as the definition of the pathways of central metabolism in generating predictive models appropriate for metabolic engineering.Another characteristic and well-known feature of plant metabolism is the extensive subcellular compartmentation that occurs within a typical plant cell (ap Rees, 1987). The cytosolic, plastidic, peroxisomal, and mitochondrial compartments are all metabolically important, with the plastids in both heterotrophic and photosynthetic cells having a notable role in biosynthesis. In some cases, particular metabolic steps occur uniquely in one compartment, for example, the synthesis of starch from ADPglucose is exclusively plastidic, but there are many instances where a particular step occurs in more than one compartment, and in extreme cases this leads to the duplication of whole pathways in two or more compartments. For example, there is considerable duplication of the pathways of carbohydrate oxidation between the cytosol and the plastids of heterotrophic tissues (Neuhaus and Emes, 2000) and many of the reactions of folate-mediated one carbon metabolism can occur in three compartments—the cytosol, mitochondria, and plastids (Hanson et al., 2000). Subcellular compartmentation has two major consequences for defining the metabolic network and constructing a predictive model of plant metabolism, and these are discussed in the following paragraphs.
First, it is necessary to identify all the transport steps that link the subcellular metabolite pools as well as the subcellular location(s) of each metabolic step. New plastidic transporters are still being identified (Weber et al., 2005), and when added to the multiple metabolite transporters in the inner mitochondrial membrane (Picault et al.,2004), the result is to add considerably to the complexity of the plant metabolic network. Moreover, identifying the subcellular location(s) of particular steps can be difficult because of the uncertainties associated with the preparation of sufficiently pure subcellular fractions from tissue extracts, and the result in any case is often both species and tissue specific. For example, the extent to which all the enzymes of the pentose phosphate pathway are present in the cytosol is variable (Debnam and Emes, 1999; Kruger and von Schaewen, 2003), and our understanding of the pathway of starch synthesis in cereal endosperm has had to be revised following the characterization of a cytosolic isoform of the normally plastidic ADPglucose pyrophosphorylase (Burton et al., 2002; Denyer et al., 1996).
Second, identical steps in different compartments are generally catalyzed by isozymes with distinct properties. Thus, duplication of pathways complicates the construction of predictive models by increasing the amount of kinetic and regulatory information that is required for the network. Moreover, the subcellular concentrations of substrates, coenzymes, and effectors will usually be different in different compartments (Farré et al., 2001), increasing the information that is required for the construction of a realistic model. A further complication is that even when an activity has been localized to a compartment, it may be distributed nonuniformly and in this situation there is the possibility that the effective concentrations of the substrates, coenzymes, and effectors will differ from their overall values. Thus, in the case of several cytosolic enzymes, there is good evidence for a membrane-associated subfraction that can be expected to have distinct kinetic properties and presumably a specific functional role within the network. Examples include nitrate reductase (Lo Piero et al., 2003; Wienkoop et al., 1999) and sucrose synthase (Amor et al., 1995; Komina et al.,2002), both of which have forms associated with the plasma membrane, and the recent demonstration of an extensive association of the enzymes of glycolysis with the outer mitochondrial membrane in Arabidopsis (Giegé et al., 2003).
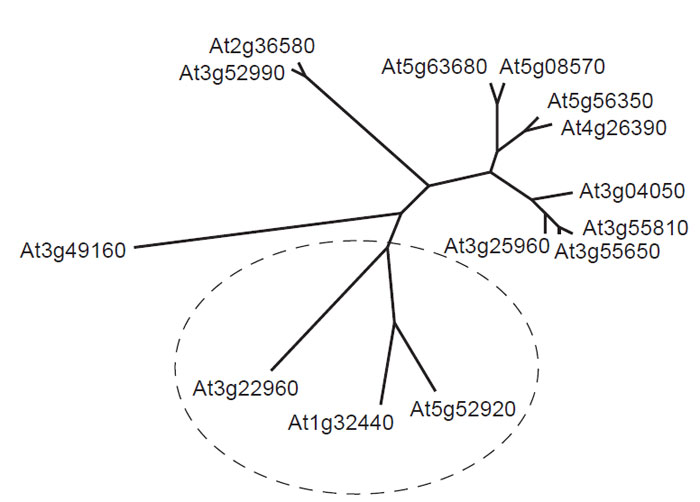 |
| FIGURE 1.1 Unrooted phylogenetic analysis of putative pyruvate kinase genes from Arabidopsis thaliana. Each gene is identified by its AGI gene code. The deduced amino acid sequences of predicted pyruvate kinase isoforms were compared using CLUSTAL W. Genes proposed to encode plastid isoforms of the enzyme were identified using ChloroP and are enclosed within the broken ellipse. Predicted transit peptides were removed prior to sequence comparison. |
Another important feature of the plant metabolic network is that much remains to be discovered before a definitive map can be drawn. This assertion is supported by the discovery of several major pathways in recent years, for example, the pathway for the synthesis of ascorbate (Smirnoff et al., 2001) and the methylerythritol pathway for the synthesis of terpenes (Eisenreich et al., 2001), and even apparently well-characterized areas of the network, such as the pathway to ADPglucose in leaves, can become candidates for reevaluation in the light of new data (Baroja- Fernandez et al., 2004, 2005; Munoz et al., 2005; Neuhaus et al., 2005). Moreover, the introduction of new techniques for probing plant metabolism invariably provides new information about the architecture and regulation of the plant metabolic network. For example, the development of insertional mutagenesis for gene silencing has generated a powerful method for probing the redundancy of the network, and this technique has been used to investigate the interaction between peroxisomes and mitochondria in plant lipid metabolism(Thorneycroft et al., 2001). There is also a very strong indication from the Arabidopsis and rice genomes that much remains to be identified before a complete metabolic network can be constructed. It is already apparent from the incompletely annotated genomes that many of the identified enzymes exist in multiple isoforms, and a notable example of this phenomenon is provided by pyruvate kinase, which appears to be represented by up to 14 genes in Arabidopsis (Fig. 1.1). Presumably different isoforms play significant roles in particular compartments of particular cell types at appropriate stages in the plant life cycle, and incorporating this level of detail into a predictive metabolic model is likely to be a major challenge.
While the complexity of the plant metabolic network is an obstacle to predictive modeling, it is also a fundamental characteristic of plant metabolism and it would be unrealistic to imagine that it can be ignored. An analysis of the metabolic network in Escherichia coli suggests that increased complexity is a desirable property for cells exposed to uncontrollable external conditions, conferring robustness and the ability to function at near optimal rates over a range of physiological conditions (Stelling et al., 2002). This fundamental property of complex systems undermines the central objective of attempting to manipulate the performance of the network through genetic engineering, and it emphasizes the importance of establishing as complete a description of the network as possible. Fortunately, annotation of the Arabidopsis and other plant genomes should provide a complete inventory of the catalytic components of various plant metabolic networks in due course, and while this will not lead to the immediate clarification of the complex relationships that determine the way in which the enzymes function in such networks, it will at least define the scale of the problem.
Assuming that the enzymes and their locations can be identified, there is still much that needs to be determined to define the metabolic network at a level that is suitable for predictive modeling of fluxes. In particular, as well as defining the levels of the enzymes and their substrates, it is also necessary to identify all the regulatory mechanisms that operate in the network. At one level, this requires the characterization of all the molecular crosstalk that allows the components of the system to influence enzyme activity through effector-binding interactions; and at a higher level, and particularly in a system that will generally not be maintained in a steady state, it is also necessary to define the relationship between gene expression and the performance of the network, for example, to include the effects of circadian rhythms, light–dark transitions, and developmental triggers on enzyme levels. Clearly, the information required to define a metabolic network at this level of precision is not available for the cells of an organism as complicated as a higher plant, and indeed it is arguable that the emerging discipline of systems biology is unlikely to provide it, since the methodological focus is analytical, concentrating on genome-scale datasets for transcripts, proteins, and metabolites rather than mechanistic (Sweetlove et al., 2003). It is also interesting to note that transcriptomic and proteomic analysis of simpler systems has not revealed direct quantitative correlations with metabolic fluxes (Oh and Liao, 2000; Oh et al>., 2002; ter Kuile and Westerhoff, 2001), demonstrating that high-throughput methods are not yet able to provide an effective alternative to the detailed kinetic and regulatory characterization of a metabolic network if the aim is predictive metabolic engineering.
While this section has emphasized the importance and difficulty of defining a complete plant metabolic network, the analysis of even an incompletely specified metabolic network can be informative. For example, genome-scale models of metabolism have been developed that allow reliable predictions of the growth potential of mutant phenotypes in E. coli, even though the analysis is based on genome annotation that is only 60–70% complete (Edwards and Palsson, 2000a; Edwards et al., 2001; Price et al., 2003). Similarly, a metabolic flux analysis of the principal pathways of carbon metabolism in Corynebacterium glutamicum was sufficiently detailed to identify a substantial diversion of resources into a cyclic flux involving the anaplerotic pathways (Petersenet al., 2000). This observation provided the basis for a rational manipulation of the system and indeed the production of a strain lacking phosphoenolpyruvate (PEP) carboxykinase had the desired effect of decreasing metabolic cycling and increasing lysine production (Petersen et al., 2001). Thus, while it is always possible that an incomplete metabolic model lacks the key feature that determines a relevant property of the system, worthwhile predictions of metabolic performance can often be made with such models. Moreover, even incorrect predictions are useful because they may suggest ways in which the model can be improved.
Support our developers




