Purification of COPI Vesicles
I. INTRODUCTIONThis article describes the in vitro assay that was developed for the faithful formation, isolation, and characterisation of COPI-derived vesicles from highly purified rat liver Golgi membranes (Lanoix et al., 1999, 2001). These vesicles normally form from Golgi cisternae carrying material from cisterna to cisterna, as well as from the cis face of the Golgi stack to the endoplasmic reticulum. Although the exact mechanism for vesicle formation and cargo incorporation is still debated, the fundamental coat components, as well as the basic principles of vesicle formation, have been clear for some time. ADP-ribosylation factor 1 (Arfl), a small GTPase of the Ras family, is first loaded with GTP via its guanine exchange factor (GEF) at the cisternal membrane. This allows Arfl to recruit coatomer (COPI) to the donor membrane. The functional cycling of coatomer on the Golgi membrane is thought to drive cargo incorporation into the forming vesicle, as well as membrane deformation and the subsequent budding of a COPI-coated vesicle. The hydrolysis of GTP by Arfl, stimulated by a GTPase-activating protein (GAP), is then thought to trigger vesicle uncoating, producing a transport intermediate capable of fusing with target membranes (Barlowe, 2000). By adding a nonhydrolysable analog of GTP to in vitro budding reactions, vesicles form but retain their coat, making them easier to isolate and analyse. The first in vitro COPI vesicle budding assays indeed relied on GTPγS for this purpose.
The following protocols are a modifications of the COPI-vesicle budding assay first described by Warren and co-workers (Sonnichsen et al., 1996). The main difference is that here we allow for GTP hydrolysis. This offers the possibilty to study vesicle formation in conditions that are more physiological by circumventing the previous need for GTPγS. Important observations have been made using this strategy. First, analysis of the protein content shows a preferential incorporation of resident proteins into COPI vesicles (Lanoix et al., 1999). Second, the mechanism of formation seems to depend on GTP hydrolysis by Arfl and cargo incorporation is linked to GAP (Lanoix et al., 1999, 2001). The coupling between GAP and cargo incorporation effectively gives rise to a kinetic proofreading mechanism ensuring that vesicles only form when sufficient and correct cargo has been selected (Weiss and Nilsson, 2003).
To summarise: Golgi stacks are purifed from rat liver and are incubated together with rat liver cytosol at 37°C. By adding the recombinant α-SNAPdn mutant to the reaction the heterotypic fusion between formed vesicles and acceptor compartments is blocked (Barnard et al., 1997). In this way only one round of vesicle formation is reconstituted. After terminating the reaction, slowly sedimenting vesicles are separated from heavier donor membranes by medium speed centrifugation. The COPI-derived vesicles are then recovered using high speed centrifugation onto a twostep sucrose cushion. Following resuspension, vesicles may then be used for a variety of biochemical or morphological purposes, such as analysis of protein or lipid content, processed for electron microscopy or used in other functional assays.
II. MATERIALS AND INSTRUMENTATION
Guanosine 5'-triphosphate lithium salt (GTP) (Cat. No. G-5884), guanosine 5'-[γ-thio]triphosphate tetralithium salt (GTPγS) (Cat. No. G-8634), benzamidine- HCl (Cat. No. B-6506), leupeptine (Cat. No. L- 2884), and soybean trypsin inhibitor (Cat. No. T-9128) are from Sigma. KOAc (Cat. No. 104820), MgCl2 (Cat. No. 105833), KCl (Cat. No. 104936), NaOH (Cat. No. 106469), and KOH (Cat. No. 105021) are from Merck. HEPES (Cat. No. 05288) and dithiothreitol (DTT, Cat. No. 4010-1) are from Biomol. ATP (Cat. No. 127523), creatine phosphate (Cat. No. 621714), and creatine kinase (Cat. No. 127566) are from Roche. Ultrapure sucrose (Cat. No. 21938) is from USB. The protein assay dye reagent (Cat. No. 500-0006) is from Bio-Rad. Bovine serum albumin (BSA, Cat. No. 23209) is from Pierce. The ECL Western blotting detection reagent (Cat. No. RPN2109) is from Amersham Biosciences. Water is of double-distilled grade (ddH2O).
Ultracentrifugation is carried out using an Optima MAX-E tabletop ultracentrifuge using a TLA-100.2 rotor (Cat. No. 362046) with polycarbonate centrifugation tubes 11 × 34mm with a 1-ml capacity (Cat. No. 343778), a TLA-55 rotor (Cat. No. 366752) with polyallomer tubes 9.5 × 38mm with a 1.5-ml capacity (Cat. No. 357448), or a Optima XL-100K ultracentrifuge using a SW-60 rotor (Cat. No. 335649) with ultraclear centrifuge tubes 11 × 60mm (Cat. No. 344062) from Beckman. Medium-speed centrifugation is carried out in a Biofuge (Cat. No. 75005510) from Heraeus using microcentrifuge tubes with a 1.5-ml capacity (Cat. No. 96.75114.9.01) from Treff lab. Round-bottom test tubes with a 14-ml capacity (Cat. No. 352006), needles 18G × 3 ½ (Cat. No. 405184), thin needles 0.4 × 19 (Cat. No. 302200), 5-ml syringes (Cat. No. 309603), and 1-ml syringes (Cat. No. 309602) are from BD Biosciences. Glass micropipettes (Cat. No. 708757) are from BRAND. The rubber adaptor between micropipettes and syringes is homemade. A standard laboratory vortex and heating block or water bath are used for incubations at 37°C. P-10, P-20, P-100, P-500, and P- 1000 pipettes are from Gilson.
III. PROCEDURES
Purification of Golgi membranes and cytosol from rat liver is described in the article by Wang et al. The concentration of the Golgi membrane aliquots should be at least 1 mg/ml and the concentration of rat liver cytosol aliquots at least 30mg/ml to accommodate the final volume of the reaction. Purification of a 10-mg/ml (approximately) stock of the His6-tagged α-SNAPdn mutant is performed as described (Barnard et al., 1997).
Protein concentration determination is performed using the Bio-Rad version of the Bradford protein assay reagent and BSA as a standard. One-dimensional SDS-polyacrylamide gel electrophforesis (SDS-PAGE) is carried out according to Laemmli (1970). Western blotting is carried out according to Towbin et al. (1979). Antibodies are revealed using chemiluminescence.
A. Small-Scale Purification
Solutions
- KOAc assay buffer: 25 mM HEPES, 115 mM KOAc, and 2.5 mM MgCl2, pH 7.0. To make 0.5 litre, dissolve 3.0g of HEPES, 5.6g of KOAc, and 0.1g of MgCl2 in 450ml water. Adjust pH to 7.0 with 1M KOH and bring to a total volume of 0.5 litre with water. Aliquot and store at -20°C.
- 10× ATP-regenerating system: 26mM of ATP, 100 mM of creatine phosphate, and 116 U/ml of creatine kinase. (A) Dissolve 0.1 g of ATP in 370 µl of KOAc assay buffer. Adjust pH to 7.0 with 1M NaOH and bring to a total volume of 700µl with KOAc assay buffer. (B) Dissolve 223 mg of creatine phosphate in a total volume of 2.3 ml KOAc assay buffer. (C) Dissolve 1 mg of creatine kinase in 333 µl of KOAc assay buffer and 333 µl of glycerol. (D) Combine the solutions and add 3.3 ml of KOAc assay buffer. Aliquot, snap freeze in liquid nitrogen, and store at -80°C.
- GTP stock solution: 40 mM GTP. Dissolve 24 mg of GTP in 1 ml of water. Aliquot and store at -20°C.
- GTPγS stock solution: 2 mM GTPγS. Dissolve 5 mg of GTPγS in a final volume of 4 ml water. Aliquot and store at -20°C.
- 100× protease inhibitor cocktail: 50mM benzamidine- HCl, 500µg/ml leupeptine, and 200µg/ml soybean trypsin inhibitor. Dissolve 39.2 mg of benzamidine-HCl, 2.5 mg of leupeptine, and 1 mg of soybean trypsin inhibitor in water to a final volume of 5 ml. Aliquot and store at -20°C.
- DTT stock solution: 100mM DTT. Dissolve 1.5 mg of DTT in 1 ml of water. Aliquot and store at -20°C.
- 3M KCl: Dissolve 22.5g KCl in 100ml water. Filter sterilise with a 0.22-µm filter and store at 4°C.
- 30% sucrose KOAc assay buffer: 25mM HEPES, 115mM KOAc, 2.5mM MgCl2, and 30% (w/w) sucrose, pH 7.0. To make 50 ml, dissolve 0.3 g HEPES, 0.56 g KOAc, 10 mg MgCl2, and 16.9 g sucrose in 45 ml of water. Adjust pH to 7.0 with 1M KOH and bring to a final volume of 50ml with water. Aliquot and store at -20°C.
- 50% sucrose KOAc assay buffer: 25mM HEPES, 115mM KOAc, 2.5mM MgCl2, and 50% (w/w) sucrose, pH 7.0. To make 50ml, dissolve 0.3 g HEPES, 0.56 g KOAc, 10mg MgCl2, and 30.7 g sucrose in 45 ml of water. Adjust pH to 7.0 with 1M KOH and bring to a final volume of 50ml with water. Aliquot and store at -20°C.
Order of operation
- Thaw 500µl of rat liver cytosol and dilute by adding 1 ml of KOAc assay buffer. Centrifuge the diluted cytosol for 1 h at 157,000 gmax at 4°C (60,000 rpm in a TLA 100.2 rotor).
- Transfer the supernatant to microcentrifuge tubes with 1.5-ml capacity and centrifuge for 15 min at 16,000g at 4°C (13,000rpm in Biofuge) to further clear the cytosol.
- Determine the protein concentration of the diluted cytosol and the Golgi membrane fraction used in the assay.
- Just before mixing the assay mixture, dilute the His6-tagged α-SNAPdn mutant to 1 mg/ml in KOAc assay buffer prewarmed to 37°C. Keep at 37°C until use.
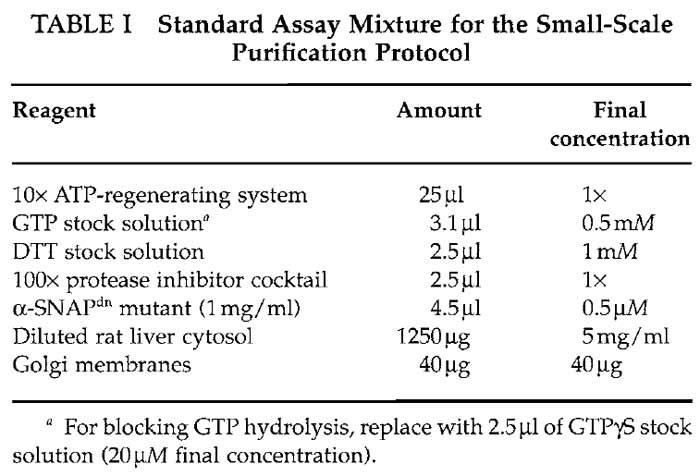
- Mix the assay mixture by adding the following reagents to a microcentrifuge tube with 1.5-ml capacity according to Table I.
- Add KOAc assay buffer so that the final assay volume is 250µl.
- Vortex the tube very briefly (not more than 1 s) to mix the assay mixture.
- Incubate for 30min at 37°C.
- Terminate the reaction by placing the tube on ice.
- To detach vesicles from membranes, raise the salt concentration to 250mM by adding 23 µl of a 3M stock solution of KCl to the reaction.
- To separate donor membranes from slowly sedimenting vesicles, centrifuge for 15min at 16,000g at 4°C (13,000rpm in a Biofuge).
- Transfer the medium-spin supernatant to a polyallomer centrifugation tube with 1.5-ml capacity without disturbing the membrane pellet.
- Create a two-step sucrose cushion by adding 50µl of 30% (w/w) sucrose KOAc assay buffer very carefully to the side of the tube, just below the surface of the supernatant followed by 5µl of 50% (w/w) sucrose KOAc assay buffer.
- To collect vesicles, centrifuge for 45min at 126,000 gmax at 4°C (45,000rpm in a TLA-55 rotor).
- Vesicles are found in the 30/50% sucrose interface. Carefully aspirate all but 20µl of the contents in the tube with a thin needle connected to a vacuum.
- For analysis of protein content by SDS-PAGE and Western blotting, add 20µl of 2× sample buffer to the tube, vortex to mix, and resuspend vesicles thoroughly by pipetting up and down with a P-20 Gilson pipette for 1 min. To ensure that all vesicles are resuspended properly, place the tube in a thermomixer and shake at maxiumum speed for 15min at 4°C. For analysis of vesicle fractions by other methods, use KOAc assay buffer instead of sample buffer.
- For analysis of protein content by SDS-PAGE and Western blotting, boil sample for 5min at 95°C and load the entire sample (40µl) onto a 1.5-mm SDS-PAGE gel. On the same gel run 1, 2, 4, and 8 gg of Golgi membranes (corresponding to 2.5, 5, 10, and 20% of starting membranes used in assay) to create a standard of Golgi proteins for analytical purposes. Analyse the protein content in the vesicle fractions using specific antibodies.
 |
B. Large-Scale Purification
Solutions (in Addition to Those Described in Section III.A)
- 20% sucrose KOAc assay buffer 0.5M KCl: 25 mM HEPES, 115 mM KOAc, 2.5 mM MgCl2, and 20% (w/w) sucrose, pH 7.0. To make 50ml, dissolve 0.3 g HEPES, 0.56 g KOAc, 10 mg MgCl2, 1.9 g KCl, and 10.8 g sucrose in 45 ml of water. Adjust pH to 7.0 with 1M KOH and bring to a final volume of 50ml with water. Aliquot and store at -20°C.
- 50% sucrose KOAc assay buffer 0.5M KCl: 25 mM HEPES, 115 mM KOAc, 2.5 mM MgCl2, and 55% (w/w) sucrose, pH 7.0. To make 50ml, dissolve 0.3 g HEPES, 0.56 g KOAc, 10 mg MgCl2, 1.9 g KCl, and 30.7 g sucrose in 45 ml of water. Adjust pH to 7.0 with 1M KOH and bring to a final volume of 50ml with water. Aliquot and store at -20°C.
- 65% sucrose KOAc assay buffer 0.5M KCl: 25 mM HEPES, 115 mM KOAc, 2.5 mM MgCl2, and 65% (w/w) sucrose, pH 7.0. To make 50ml, dissolve 0.3 g HEPES, 0.56 g KOAc, 10 mg MgCl2, 1.9 g KCl, and 42.8 g sucrose in 45 ml of water. Adjust pH to 7.0 with 1M KOH and bring to a final volume of 50ml with water. Aliquot and store at -20°C.
Order of operation
- Thaw 2.5 ml of rat liver cytosol and dilute it by adding 5ml of KOAc assay buffer. Centrifuge the diluted cytosol for 1 h at 157,000 gmax at 4°C (60,000 rpm in a TLA 100.2 rotor).
- Transfer the supernatant to microcentrifuge tubes with 1.5-ml capacity and centrifuge for 15 min at 16,000g at 4°C (13,000rpm in Biofuge) to further clear the cytosol.
- Pool supernatants and determine the protein concentration of the diluted cytosol and the Golgi membrane fractions used in the assay.
- Just before mixing the assay mixture, dilute the His6-tagged α-SNAPdn mutant to 1 mg/ml in KOAc assay buffer prewarmed to 37°C. Keep at 37°C until use.
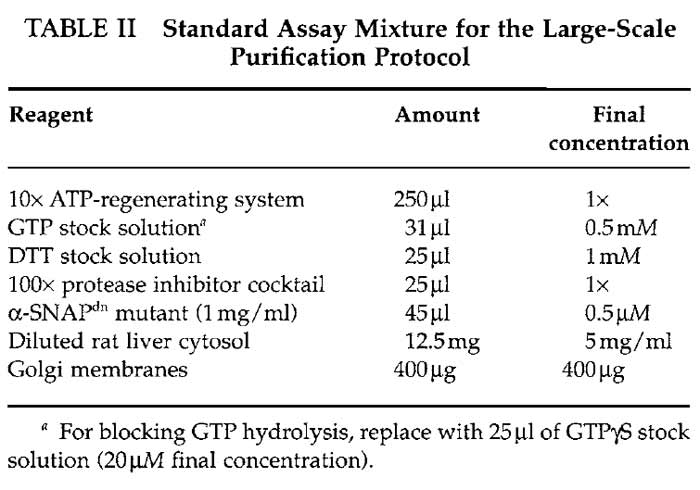
- Mix five standard assay mixtures by adding the following reagents to five round-bottom test tubes with 14-ml capacity according to Table II.
- Add KOAc assay buffer up to 2.5 ml final reaction volume in each tube.
- Vortex the tubes very briefly (not more than 1 s) to mix the assay mixture.
- Incubate for 30min at 37°C.
- Terminate the reaction by placing tubes on ice.
- To detach vesicles from membranes, raise the salt concentration to 250mM by adding 230 µl of a 3 M stock solution of KCl to each of the five tubes.
- To separate donor membranes from slowly sedimenting vesicles, divide the content of each tube into 0.3-ml fractions into several (around 40 in total)microcentrifuge tubes with 1.5-ml capacity and centrifuge for 15min at 16,000g at 4°C (13,000rpm in Biofuge).
- Pool the medium spin supernatants (around 12ml in total volume) in a round-bottom test tube without disturbing the membrane pellets.
- Prepare four two-step sucrose cushions by adding 800µl of 20% (w/w) sucrose KOAc assay buffer 0.5M KCl to an ultraclear SW-60 centrifuge tube and then underlay this with 150µl of 50% (w/w) sucrose KOAc assay buffer 0.5M KCl using a micropipette connected to a 1-ml syringe. Onto each gradient overlay 3 ml of the pooled supernatant using a 5-ml syringe connected to a needle.
- To collect vesicles, centrifuge for 3 h at 407,000 gmax at 4°C (55,000rpm in a SW-60 rotor).
- Vesicles are found in the 20/50% sucrose interface. Carefully aspirate all but 500µl of the contents in the tube with a thin needle connected to a vacuum.
- Collect the vesicles by removing 300 µl from the bottom of each tube using a micropipette connected to a 1-ml syringe.
- Pool these vesicle fractions into a new roundbottom test tube and adjust the sucrose concentration to approximately 55% sucrose (w/w) KOAc assay buffer 0.5M KCl by adding 1550 µl 65% sucrose (w/w) KOAc assay buffer 0.5 M KCl.
- To further purify vesicles by floatation, prepare
a step sucrose gradient in an ultraclear SW-60 centrifuge
tube. Start with the 20% sucrose (w/w) KOAc
assay buffer 0.5 M KCl. This is underlaid with 50%
sucrose (w/w) KOAc assay buffer, which is underlaid
with the sample containing the vesicles in 55% sucrose
using a micropipette connected to a syringe in the following
order:
- 500µl 20% sucrose (w/w) KOAc assay buffer 0.5 M KCl
- 750µl 50% sucrose (w/w) KOAc assay buffer 0.5 M KCl
- 2750µl 55% sucrose (w/w) KOAc assay buffer 0.5 M KCl sample from previous step.
- To float vesicles, centrifuge for 16h at 337,000 gmax at 4°C (50,000rpm in a SW-60 rotor).
- Collect vesicles by removing 100-µl fractions from the top of the gradient. The layer of vesicles at the interface is between 20/50% sucrose.
 |
IV. Comments
The small-scale protocol is useful for comparing a variety of reaction conditions in one experiment. The recovery of various proteins in the vesicle fractions is compared to the amount of protein in the protein standard on the gel. For any condition that is tested it is strongly recommended to always run controls containing GTP and GTPTS and one at 4~ Comparing these three control conditions with the one tested usually gives a good indication of the effect that the particular condition has. The large-scale protocol gives a higher amount of vesicles and is more suitable when more material is needed, for example protein analysis through proteomics.
V. PITFALLS
- Unless specified, all steps should be carried out on ice to minimise protein degradation. Aliquots for one-time use should be thawed and kept on ice until use.
- Pipetting of Golgi membranes should be minimised to avoid shearing. Mix these aliquots by tapping carefully on the side of the tube.
- Once the protein concentration of a certain batch of purifed Golgi membranes and cytosol has been established, it is not necessary to perform the protein concentration determination in step 3 of the protocols.
- The sucrose content of the various buffers should be verified with a refractometer.
- Aspiration steps may represent a source of variation. Take special care during these steps.
Acknowledgments
The authors thank Dr. Joel Lanoix for developing this assay and Nilsson group members for providing critical comments of the manuscript.
References
Barlowe, C. (2000). Traffic COPs of the early secretory pathway. Traffic 1, 371-377.
Barnard, R. J., Morgan, A., and Burgoyne, R. D. (1997). Stimulation of NSF ATPase activity by alpha-SNAP is required for SNARE complex disassembly and exocytosis. J. Cell Biol. 139, 875-883.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685.
Lanoix, J., Ouwendijk, J., Lin, C. C., Stark, A., Love, H. D., Ostermann, J., and Nilsson, T. (1999). GTP hydrolysis by arf-1 mediates sorting and concentration of Golgi resident enzymes into functional COP I vesicles. EMBO J. 18, 4935-4948.
Lanoix, J., Ouwendijk, J., Stark, A., Szafer, E., Cassel, D., Dejgaard, K., Weiss, M., and Nilsson, T. (2001). Sorting of Golgi resident proteins into different subpopulations of COPI vesicles: A role for ArfGAP1. J. Cell Biol. 155, 1199-1212.
Sonnichsen, B., Watson, R., Clausen, H., Misteli, T., and Warren, G. (1996). Sorting by COP I-coated vesicles under interphase and mitotic conditions. J. Cell Biol. 134, 1411-1425.
Towbin, H., Staehelin, T., and Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350-4354.
Weiss, M., and Nilsson, T. (2003). A kinetic proof-reading mechanism for protein sorting. Traffic 4, 65-73.
Support our developers




