Hemostasis: Prevention of Blood Loss
Hemostasis: Prevention
of Blood Loss
It is essential that animals have ways of
preventing rapid loss of body fluids
after an injury. Since blood is flowing
and is under considerable hydrostatic
pressure, it is especially vulnerable to
hemorrhagic loss.
When a vessel is damaged, smooth muscle in the wall of the vessel contracts, which causes the vessel lumen to narrow, sometimes so strongly that blood flow is completely stopped. This simple but highly effective means of preventing hemorrhage is used by invertebrates and vertebrates alike. Beyond this first defense against blood loss, all vertebrates, as well as some larger, active invertebrates with high blood pressures, have in the blood special cellular elements and proteins that are capable of forming plugs, or clots, at the injury site.
In vertebrates blood coagulation is the dominant hemostatic defense. Blood clots form as a tangled network of fibers from one of the plasma proteins, fibrinogen. The transformation of fibrinogen into a fibrin meshwork (Figure 33-5) that entangles blood cells to form a gel-like clot is catalyzed by the enzyme thrombin. Thrombin is normally present in blood in an inactive form called prothrombin, which must be activated for coagulation to occur.
In this process, blood platelets (Figure 33-3) play a vital role. Platelets form in red bone marrow from certain large cells that regularly pinch off bits of their cytoplasm; thus they are fragments of cells. There are 150,000 to 300,000 platelets per cubic millimeter of blood. When the normally smooth inner surface of a blood vessel is disrupted, either by a break or by deposits of a cholesterol-lipid material, platelets rapidly adhere to the surface and release thromboplastin and other clotting factors. These factors, along with factors released from damaged tissue and with calcium ions, initiate conversion of prothrombin to active thrombin (Figure 33-6).
The catalytic sequence in this scheme is unexpectedly complex, involving a series of plasma protein factors, each normally inactive until activated by a previous factor in the sequence. The sequence behaves like a “cascade” with each reactant in the sequence leading to a large increase in the amount of the next reactant. At least 13 different plasma coagulation factors have been identified. A deficiency of only a single factor can delay or prevent the clotting process. Why has such a complex clotting mechanism evolved? Probably it is necessary to provide a fail-safe system capable of responding to any kind of internal or external hemorrhage that might occur and yet a system that cannot be activated into forming dangerous intravascular clots in the absence of injury.
Several kinds of clotting abnormalities in humans are known. One of these, hemophilia is a condition characterized by failure of blood to clot, so that even insignificant wounds can cause continuous severe bleeding. It is caused by a rare mutation (the condition occurs in about 1 in 10,000 males) on the X sex chromosome, resulting in an inherited lack of one of the platelet factors in males and in homozygous females. Called the “disease of kings,” it once ran through several interrelated royal families of Europe, apparently having originated from a mutation in one of Queen Victoria’s parents.
Hemophilia is one of the best known cases of sex-linked inheritance in humans. Actually two different loci on the X chromosome are involved. Classic hemophilia (hemophilia A) accounts for about 80% of persons with the condition, and the remainder are caused by Christmas disease (hemophilia B).The allele at each locus results in a deficiency of a different platelet factor.
 |
| Figure 33-5 Human red blood cells trapped in fibrin clot. Clotting is initiated after tissue damage by disintegration of platelets in blood, resulting in a complex series of intravascular reactions that end with conversion of a plasma protein, fibrinogen , into long, tough, insoluble polymers of fibrin. Fibrin and entangled erythrocytes form the blood clot, which arrests bleeding. |
When a vessel is damaged, smooth muscle in the wall of the vessel contracts, which causes the vessel lumen to narrow, sometimes so strongly that blood flow is completely stopped. This simple but highly effective means of preventing hemorrhage is used by invertebrates and vertebrates alike. Beyond this first defense against blood loss, all vertebrates, as well as some larger, active invertebrates with high blood pressures, have in the blood special cellular elements and proteins that are capable of forming plugs, or clots, at the injury site.
In vertebrates blood coagulation is the dominant hemostatic defense. Blood clots form as a tangled network of fibers from one of the plasma proteins, fibrinogen. The transformation of fibrinogen into a fibrin meshwork (Figure 33-5) that entangles blood cells to form a gel-like clot is catalyzed by the enzyme thrombin. Thrombin is normally present in blood in an inactive form called prothrombin, which must be activated for coagulation to occur.
In this process, blood platelets (Figure 33-3) play a vital role. Platelets form in red bone marrow from certain large cells that regularly pinch off bits of their cytoplasm; thus they are fragments of cells. There are 150,000 to 300,000 platelets per cubic millimeter of blood. When the normally smooth inner surface of a blood vessel is disrupted, either by a break or by deposits of a cholesterol-lipid material, platelets rapidly adhere to the surface and release thromboplastin and other clotting factors. These factors, along with factors released from damaged tissue and with calcium ions, initiate conversion of prothrombin to active thrombin (Figure 33-6).
The catalytic sequence in this scheme is unexpectedly complex, involving a series of plasma protein factors, each normally inactive until activated by a previous factor in the sequence. The sequence behaves like a “cascade” with each reactant in the sequence leading to a large increase in the amount of the next reactant. At least 13 different plasma coagulation factors have been identified. A deficiency of only a single factor can delay or prevent the clotting process. Why has such a complex clotting mechanism evolved? Probably it is necessary to provide a fail-safe system capable of responding to any kind of internal or external hemorrhage that might occur and yet a system that cannot be activated into forming dangerous intravascular clots in the absence of injury.
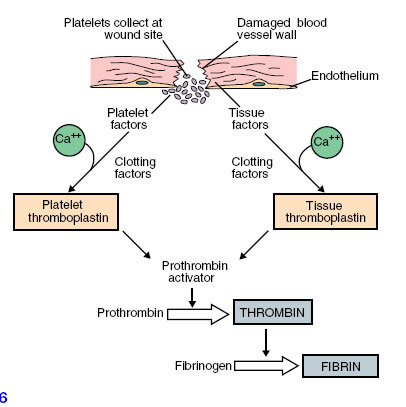 |
| Figure 33-6 Stages in formation of fibrin. |
Several kinds of clotting abnormalities in humans are known. One of these, hemophilia is a condition characterized by failure of blood to clot, so that even insignificant wounds can cause continuous severe bleeding. It is caused by a rare mutation (the condition occurs in about 1 in 10,000 males) on the X sex chromosome, resulting in an inherited lack of one of the platelet factors in males and in homozygous females. Called the “disease of kings,” it once ran through several interrelated royal families of Europe, apparently having originated from a mutation in one of Queen Victoria’s parents.
Hemophilia is one of the best known cases of sex-linked inheritance in humans. Actually two different loci on the X chromosome are involved. Classic hemophilia (hemophilia A) accounts for about 80% of persons with the condition, and the remainder are caused by Christmas disease (hemophilia B).The allele at each locus results in a deficiency of a different platelet factor.
Support our developers




