Mapping Cloned DNA on Metaphase Chromosomes Using Fluorescence in Situ Hybridization
I. INTRODUCTIONFluorescence in situ hybridization (FISH) provides a quick means of placing almost any recombinant DNA clone onto a physical map. Clones can be mapped onto banded metaphase chromosomes for regional localization, to assign new markers to a chromosomal segment, to validate clones selected using previously assigned markers, or to characterize chromosomal rearrangements.
In genome mapping, unrecognized clone chimerism causes considerable problems with contig assembly and can result in much wasted effort when trying to extend or link contigs. For example, as many as 30-40% of YAC clones may contain coligated sequences from different genomic regions (Selleri et al., 1992). The shift to large-insert, single-copy plasmids such as PACs and BACs has substantially reduced the problem of clone chimerism. However, large-scale genome sequencing projects are revealing significant levels of regional homology that can confound mapping analyses at all levels (Cheung et al., 2001; Bailey et al., 2002). Preliminary FISH screening of clones is a simple and useful precaution.
DNA clones can also be ordered by FISH at increasing levels of resolution. Clones separated by 1-2Mb can be ordered by pairwise, two-color FISH on metaphase chromosomes (Trask et al., 1991; Inazawa et al., 1994). Hybridization of sets of three clones to interphase nuclei provides mapping information down to about 50-100kb (Lawrence et al., 1990; Trask et al., 1991), while the highest resolution is for overlapping clones on linear DNA molecules (Raap et al., 1996).
II. MATERIALS AND INSTRUMENTATION
DNase I (D-4527), DNA polymerase I (D-9380), bovine serum albumin (BSA, B-4287), dextran sulfate (D-8906), Denhardt's solution (D-2532), dithiothreitol (DTT, D-9779), SDS or sodium lauryl sulfate (L-4390), mouse antidigoxin FITC (F-3523), goat anti-mouse- FITC (F-0257), rabbit anti-mouse-FITC (F-7506), and DAPI (D-9542) are from Sigma. Deoxyribonucleotides (100mM solutions of dATP, dCTP, dGTP, and dTTP) are from Amersham Biosciences (Cat. No. 27-2035-01). Human Cot-1 DNA (Cat. No. 15279011) is from Invitrogen. Formamide is from Fluka (Cat. No. 47670). Molecular biology grade mixed bed resin AG 501-X8 is from Bio-Rad (Cat. No. 143-6424). Biotin-16-dUTP (Cat. No. 1093070) and digoxigenin-11-dUTP (Cat. No. 1573152) are from Roche Diagnostics.
Berliner Glas microscope slides are from H. V. Skan Ltd., Solihull. Glass coverslips (No. 1-22 × 22mm, 22 × 32mm, 22 × 50mm) and Nescofilm are from Fisher Scientific. Rubber cement can be obtained from art suppliers or from cycle shops (e.g., Halfords). Plastic syringes are from Becton-Dickinson, and 0.2-µm syringe filters are from Nalgene (Cat. No. 190-2520). Plastic slide boxes (50 slide capacity, Cat. No. 406028600), self-indicating silica gel (Cat. No. 300624V), Tween 20 (Cat. No. 66368 4B), glycerol (Cat. No. 101186M), 96% ethanol, and 99% ethanol are from BDH (VWR International).
Fine forceps suitable for handling slides and coverslips comfortably, without mishap (13mm, Cat. No. E12), glass Coplin jars (Cat. No. E94), Hellendahl jars (Cat. No. E95), slide racks (Cat. No. E89.03, E89.055), and diamond marking pencils (Cat. No. E17) are from Raymond A. Lamb.
Cy3-streptavidin (Cat. No. PA43001) is from Amersham Biosciences. Texas red avidin DCS (Cat. No. A- 2016), biotinylated antiavidin (Cat. No. BA-0300), and Vectashield (Cat. No. H-1000) are from Vector Laboratories Ltd. Filter paper discs, grade 4 (Cat. No. 1004 240), are from Whatman. Nonfat milk powder (e.g., Marvel) is widely available. Similarly, nail varnish from any inexpensive source should be adequate.
DNA concentrations are determined using a Hoefer TKO minifluorimeter. The slide drying bench (Cat. No. E18.1) is from Raymond A. Lamb. Also needed are at least two water baths (to be set at 37 and 65°C), a microcentrifuge for both 0.5- and 1.5-ml tubes, a 37°C oven, and facilities for performing agarose gel electrophoresis, as well as countup/countdown timers (e.g., Smiths), thermometers, micropipettes and sterile tips, domestic air-tight plastic freezer boxes, and lintfree tissues (Kimwipes). Incubation chambers are prepared using 245 × 245-mm2 bioassay dishes (Nunc, Cat. No. 240835). Racks to accommodate a maximum of 16 slides are made by trimming four plastic 10-ml pipettes to fit and fixing them in place with rubber cement.
We use a Zeiss Axioskop epifluorescence microscope fitted with a 100-W mercury arc lamp, triple bandpass/dichroic filter blocks for both rhodamine (or Cy3) and Texas red (Chroma Technologies, sets 82000 and 83000), and a motorized excitation filter wheel (Ludl) and a Photometrics KAF1400 cooled CCD camera controlled by SmartCapture imaging software. Similar imaging systems can be obtained from Applied Imaging. SmartCapture software is supplied by Digital Scientific.
III. PROCEDURES
A. Probe Labeling
Solutions
- 10× nick translation buffer: 0.5 M Tris-HCl, pH 7.5, 0.1M MgSO4, 1mM DTT, and 500 µg/ml BSA. To make 10 ml, mix 5 ml 1M Tris-HCl (pH 7.5), 1ml 1M MgSO4, 10µl 1M DTT, 500µl 10mg/ml BSA, and make up to 10ml with sterile deionized water. Store 1-ml aliquots at -20°C.
- DNase I stock solution: Resuspend 10,000 units in 1ml 0.3 M NaCl and add 1ml sterile glycerol. Store at -20°C.
- DNase I working solution: To make 1ml, mix 100 µl 10x nick translation buffer with 400 µl sterile deionized water and 500µl sterile glycerol; then add 1µl DNase I stock solution. Mix thoroughly and store at -20°C.
- 0.5 mM dNTPs: To make 1200µl, mix 2 µl each of 100mM dATP, dCTP, and dGTP and then add 1194µl sterile deionized water. Store 50-µl aliquots at -20°C.
- 1mM biotin-16-dUTP or digoxigenin-11-dUTP
- DNA polymerase I (10 units/µl)
- 0.5M EDTA, pH 8.0: To make 100ml, mix 18.61 g EDTA in 80ml deionized water, adjust to pH 8.0 with NaOH (the salt will not dissolve until near pH 8), and make up to volume with deionized water. Sterilize by autoclaving.
- 1M Tris-Cl, pH 7.4: To make 100ml, dissolve 12.11 g Tris base in 80ml deionized water, adjust to pH 7.4 with HCl, and make up to volume with deionized water. Sterilize by autoclaving.
- 3M sodium acetate, pH 7.0: To make 100ml, dissolve 24.6g sodium acetate (anhydrous) in 80ml deionized water, adjust to pH 7.0 with glacial acetic acid, and make up to 100ml with deionized water. Sterilize by autoclaving. Use small aliquots as working stock, replacing frequently.
- Absolute ethanol: Store in a sterile 50-ml Falcon tube at -20°C.
- 70% ethanol: Store in a sterile 50-ml Falcon tube at -20°C.
- TE buffer (10mM Tris-Cl, 1mM EDTA): To make 100ml, mix 1ml of 1M Tris-Cl, pH 7.4, and 0.2ml of 0.5M EDTA and make up to 100ml with deionized water. This is best made prior to sterilizing the stock solutions of Tris and EDTA. Sterilize by autoclaving.
Steps
- Prepare a water bath at 14°C. A robust polystyrene box (or ice bucket) with a lid is a suitable container (although some are not reliably water tight).
- Place approximately 1 µg of each DNA sample to be labeled in 1.5-ml microfuge tubes and make up the volumes to 10µl with sterile deionized water. Stand on ice.
- Prepare 15µl labeling master mix for each sample, allowing a little extra for dispensing. For 4 µg DNA, take 10µl nick translation buffer, add 34µl sterile deionized water, 7.5µl dNTPs, 2.5µl 1mM biotin-16-dUTP, 4µl DNase I working solution, and 2µl 10 U/µl DNA polymerase I. Mix thoroughly and pulse microfuge to collect all the solution in the base of the tube. Stand on ice.
- Add 15 µl of master mix to each DNA sample and mix by pipetting several times.
- Incubate the samples at 14°C for 40-60min. The exact length of time must be determined for each new preparation of DNase I working solution.
- Transfer the tubes to ice and add 2.5µl 0.5M EDTA to each, mixing quickly with the pipette.
- Add 2.5 µl 3M sodium acetate and 1ml cold 100% ethanol to each tube. Mix by inversion.
- Incubate at -20°C overnight.
- Microfuge the tubes for 10min at maximum speed. Remove the supernatants carefully so as not to disturb the visible pellets.
- Carefully add 1ml cold 70% ethanol to each tube. Microfuge again for 10 min. Carefully aspirate off the supernatant from the transparent pellets and air dry. Do not overdry.
- Add 10µl TE buffer to each tube and stand on ice for 15 min. Flick mix to resuspend.
- Run 2-µl aliquots in a 1% agarose gel to assess the probe fragment length. A smear between 200 and 800bp is satisfactory.
- Store the probes at -20°C.
B. in situ Hybridization
Solutions
- 50% dextran sulfate: To make 50ml, weigh 25g dextran sulfate into a graduated 100-ml pyrex bottle, add 20ml deionized water, and heat in a 65°C water bath until dissolved. Make up to volume with deionized water and sterilize by autoclaving. Dispense 10-ml aliquots into sterile 50-ml Falcon tubes and store at -20°C.
- Deionized formamide: Add 5 g Bio-Rad mixed bed resin AG 501-X8 to 100ml formamide and stir in a fume hood for 60min. Allow the beads to settle and decant the formamide. Store aliquots at -20°C.
- 20× SSC (3 M NaCl, 0.3 M sodium citrate): To make 1 liter, dissolve 175.2g sodium chloride and 88.2g trisodium citrate in deionized water and make up to volume.
- 10% SDS: To make 50ml, dissolve 5g sodium dodecyl sulfate in 50ml deionized water. Sterilize by filtering through a 0.2-µm filter.
- 0.5M Na2HPO4: To make 100 ml, dissolve 7.098 g Na2HPO4 in deionized water and make up to volume. Sterilize working aliquots by filtration.
- 0.5M NaH2PO4: To make 100ml, dissolve 7.8g NaH2PO4.2H2O in deionized water and make up to volume. Sterilize working aliquots by filtration.
- Hybridization buffer: To make 50 ml, thaw a Falcon tube containing a 10-ml aliquot of 50% dextran sulfate, add 25 ml deionized formamide, 5 ml 20× SSC, 1ml 50x Denhardt's solution, 4ml 0.5M sodium phosphate buffer, pH 7.0 (2.308 ml Na2HPO4, 1.692ml NaH2PO4), 0.5 ml10% SDS, and 4.5 ml sterile deionized water. Mix very thoroughly by inversion, preferably on a rotator for 10min. Dispense into 1-ml aliquots and store at -20°C. This should be stable for at least 1 year. Mix newly thawed aliquots thoroughly by vortexing.
- Cot-1 DNA: Supplied at 1mg/ml.
- 70% formamide: To make 100 ml, mix 70 ml formamide and 30ml 2× SSC. Store at 4°C between uses and replace weekly. (Deionizing the formamide is not necessary.)
- 70% ethanol in a Hellendahl jar: Store at -20°C. Replace weekly.
- Ethanol series: Hellendahl jars containing 70% ethanol (2×), 90% ethanol (2×), and 100% ethanol (1×).
- Coverslips: 22 × 22-mm, 22 × 50-mm coverslips immersed in 100% ethanol in an air-tight plastic container.
Steps
- Prepare a Coplin jar or Hellendahl jar containing 70% formamide. Place in a water bath and turn the temperature to 65°C. (The glass jars will not tolerate sudden temperature changes and should always be allowed to warm gradually.)
- Set a second water bath to 37°C.
- Prepare the probe hybridization mixes by adding 10µl hybridization buffer to 0.5-ml microfuge tubes (dispensing will be aided by first warming the hybridization buffer). Stand the tubes on ice. Add I or 2µl Cot-1 DNA to each tube, depending on whether one or two probes are to be added. Add 0.5µl of biotinylated probe and/or 0.5µl of another digoxigenin- labeled probe. Mix thoroughly by flicking the tubes and pulse microfuge the solutions to the bottom of the tubes.
- Denature the probe mixes at 65°C for 10min, ensuring that the tubes are fully sealed and not likely to take up water.
- Transfer the probe mixes to the 37°C water bath to preanneal for at least 20min (to several hours).
- Check that the 70% formamide has reached 65°C using a thermometer reserved for the purpose.
- Carefully immerse slides, paired back to back, into the 70% formamide at 5-s intervals. Start a countup timer as the first pair of slides is immersed. Take care to hold the slides clear of the steam from the water bath while preparing to place them in the formamide.
- While the slides are denaturing, remove the Hellendahl jar of 70% ethanol from the freezer.
- Denature the slides for 2min. As the denaturation time elapses, remove the pairs of slides from the formamide and drain briefly against the inside of the jar. Immerse the slides, agitating briefly, in the cold 70% ethanol.
- After 60s in the cold 70% ethanol, transfer the slides with occasional agitation through an ethanol series at room temperature: 60 s each in successive jars of 70, 70, 90, 90, and 100% ethanol.
- Separate the slide pairs and carefully wipe the backs of the slides dry. Air dry the slides standing in a rack on a slide-warming bench at 37-40°C.
- Prepare the required number of 22 × 22-mm coverslips by removing them from the 100% ethanol and lightly polishing them dry with a lint-free tissue. Place the coverslips on a clean tissue on or by the slidewarming bench.
- Place the dry slides flat on the slide-warming bench. Make sure that all are labeled and numbered clearly.
- After the probes have preannealed at 37°C for at least 20min, transfer the first hybridization mix to its target slide. As the volumes can be difficult to gauge accurately, set the micropipette for 12 µl and be careful to avoid bubbles when drawing up and discharging the mix onto the slide.
- Using fine forceps, gently lower a clean coverslip over the mix. If bubbles are present, these can usually be disrupted by surface tension by allowing the hybridization mix to spread out under the upper half of the coverslip while the lower edge of the coverslip is still supported by the tip of the forceps. It is usually not worth trying to remove every last bubble as this may damage the chromosome preparations.
- Repeat these steps for each remaining probe mix. If there are a large number to be processed, it may be preferable to station the slide bench next to the 37°C water bath and remove each mix from the water bath as required. Alternatively, the hybridization mixes can be placed on ice, but this can make the mixes more viscous and difficult to pipette.
- Seal the edges of the coverslips with rubber cement. If the hybridization mix has not reached the edges of a coverslip because the coverslip cannot lie flat, it will be necessary to prevent the rubber cement from being drawn under the coverslip. If the coverslip cannot be flattened by gentle pressure over the high point, fill in the gap with extra hybridization buffer before sealing.
- Place on a tray and hybridize overnight in a 37°C oven.
C. Visualization of Hybridization
Solutions
- 50% formamide: To make 200 ml, mix 100 ml formamide and 100ml 2× SSC. Store at 4°C between uses and replace weekly.
- 2× SSC: To make 1 liter, take 100ml 20x SSC and make up to volume with deionized water.
- 4 × TNFM: To make 1 liter, take 200ml 20x SSC, add 700ml deionized water, 500 µl Tween 20, and 50 g nonfat milk powder and mix vigorously, then make up to 1 liter with deionized water. Filter through several layers of Whatman No. 4 filter paper. (The solution should be a slightly translucent yellow-green color. If it remains cloudy try another brand of nonfat milk powder.)
- Immunochemical solutions: Allow 100µl per slide
plus 50-100µl excess. Protect from strong light.
- For biotinylated probes, make 2 aliquots of 4µg/ml streptavidin-Cy3 and one aliquot of 4 µg/ml biotinylated antiavidin.
- For digoxigenin-labeled probes, make a 1:500-1:1000 dilution of mouse antidigoxin-FITC and a 1:250 dilution of rabbit anti-mouse-FITC.
- For dual-color detection, use avidin-Texas red instead of streptavidin-Cy3. Combine the biotinylated antiavidin with the mouse antidigoxin-FITC, and the second avidin- Texas red with the rabbit anti-mouse-FITC.
- 4 × T: To make 200 ml, mix 40 ml 20x SSC, 160 ml deionized water, and 100µl Tween 20.
- DAPI staining solution: Prepare a Hellendahl jar with 75ml of 2× SSC and 6µl 1mg/ml DAPI. Protect from light by wrapping in aluminium foil.
Steps
- Place three Coplin jars or Hellendahl jars containing 2× SSC and two jars of 50% formamide into a water bath and warm to 42°C.
- Place a jar of 4 × TNFM to warm in a 37°C oven.
- Prepare a humidified chamber by placing damp tissues in the bottom of an incubation dish and warm in a 37°C oven.
- Prepare immunochemical solutions diluted in 4 x TNFM according to the haptens used. Stand at room temperature for 10min and then microfuge for 10min to pellet any protein complexes that might contribute to nonspecific background.
- Remove the rubber cement from the slides using fine forceps and soak off the coverslips in the first jar of 2× SSC at 42°C, allowing approximately 5 min.
- Transfer the slides to the first jar of 50% formamide for 5 min.
- Repeat this incubation in the second jar of 50% formamide and then in each of the remaining jars of 2× SSC, agitating the slides briefly after each transfer.
- Transfer the slides to the prewarmed jar of 4 x TNFM and incubate for 5-10min at 37°C.
- Remove the slides one by one from the 4 × TNFM and drain briefly, wipe the back, and blot excess liquid from the top and bottom edges. Place the slides on a rack in the prepared humidified chamber and apply 100µl of the first immunochemical solution (streptavidin- Cy3 or mouse antidigoxin-FITC or avidin-Texas red). Overlay with a 25 × 50-mm strip of Nescofilm. Note: The slides must not dry out at any point during the procedure.
- Incubate the slides at 37°C for 20-30min.
- Meanwhile, discard the three 2× SSC wash solutions and replace with 4 × TNFM, allowing the jars to warm to 42°C again.
- Discard the Nescofilm strips, drain the slides, and rinse them in the three changes of 4 × TNFM at 42°C for 5 min each.
- Repeat step 9 using the second antibody layer (biotinylated antiavidin, rabbit anti-mouse-FITC, or biotinylated antiavidin plus mouse antidigoxin-FITC).
- Repeat steps 10-12.
- Repeat step 9 using the final immunochemical layer (streptavidin-Cy3 or avidin-Texas red plus rabbit anti-mouse-FITC). If using only digoxigenin-labeled probes, proceed to step 17.
- Repeat steps 10-12.
- Wash twice in 4 × T at room temperature.
- Stain the slides in DAPI for 2-3 min. Transfer the slides to a jar containing 2× SSC, rinse briefly, and pour off the 2× SSC (holding the slides in place with a gloved finger or forceps across the top of the jar). Rinse briefly with deionized water and pour off.
- Dehydrate the slides by passing through an ethanol series, gently agitating 30-60 s in each of 70, 70, 90, 90, and 100% ethanol. Air dry.
- Lightly polish 22 × 50-mm coverslips and lay them out on flat absorbant tissue. Apply 25-30µl antifade solution to each coverslip.
- Invert each slide over a coverslip, rest the bottom edge on the tissue, and gently lower until the slide touches the antifade droplet. Allow the coverslip to lift up to the slide before laying it flat. When the antifade has spread fully, gently blot any excess. Seal the edges of the coverslips with nail varnish. The slides can now be stored in the dark at 4°C for many weeks.
- View the slides using an epifluorescence microscope equipped with the excitation and emission filters appropriate for the fluorochromes used.
IV. COMMENTS
A. Probe Labeling
Whole clone DNA preparations are usually the best material for nick translation. Bacterial clone DNA isolated by standard alkaline lysis should be satisfactory. Most matrix-binding protocols, as used by commercial kits, do not give very high yields for larger insert clones such as cosmids, PACs, or BACs (often only enough for one or two labeling reactions from 10 ml of bacterial culture) so be sure that sufficient DNA (of a concentration of at least 100µg/ml) is obtained. Total yeast DNA gives good results for YACs, although the yeast genome may contribute excess ribosomal sequences that may not be fully suppressed during hybridization.
Different grades of DNase 1 have different levels of activity. New working solutions should be titrated to determine the best incubation time. A 50-µl reaction containing 2µg DNA and 2µl DNase I working solution in 1× nick translation buffer can be sampled at five intervals (e.g., at 20, 30, 40, 50, and 60min) and the DNA fragments compared by electrophoresis. Even less expensive products, such as DN-25 (Sigma), are suitable for use after titration, allowing for the relative number of units per milligram of protein in the working solution. Enzyme activity is also affected by the amount of Ca2+ present in the reaction. Usually, sufficient Ca2+ is present in the original DNase I stock; however, higher grades of enzyme may need additional CaCl2 to maintain active enzyme conformation during the nick translation reaction.
Approximately 70-80% of the original DNA is usually recovered after ethanol precipitation, giving a final probe concentration of 70-80ng/µl. Probe concentrations can be verified by DNA fluorimetry if desired. Ethanol precipitation is not absolutely required for probe preparation and can be omitted after labeling repetitive DNA clones, such as chromosome- specific satellite DNAs, as these are routinely used at much lower hybridization concentrations (0.5-1ng/µl) than "unique-sequence" clones (2.5- 5ng/µl). Ethanol precipitation enables the labeled probe to be concentrated so that it can be used without further precipitation prior to hybridization. This ensures more consistent results from one experiment to another.
Large insert clones can be mapped efficiently using direct labeling with fluorochrome-conjugated dUTPs. This avoids the time-consuming immunochemical detection steps described here, but may not be as effective in revealing smaller signals at secondary chromosomal sites.
B. Hybridization
The hybridization buffer can be dispensed more accurately after it is warmed to 65°C. The components may separate during freezing and the warmed solution should be mixed thoroughly before use. Probes will last several years if handled aseptically and stored at -20°C. They should be thawed and kept on ice when in use and returned to the freezer without delay (do not store in frost-free freezers). Routine metaphase mapping throughput can be increased by using pairs of biotin- and digoxigenin-labeled probes on two separate spots of metaphase cells on a single slide. The slide can be mounted in antifade under a single large coverslip.
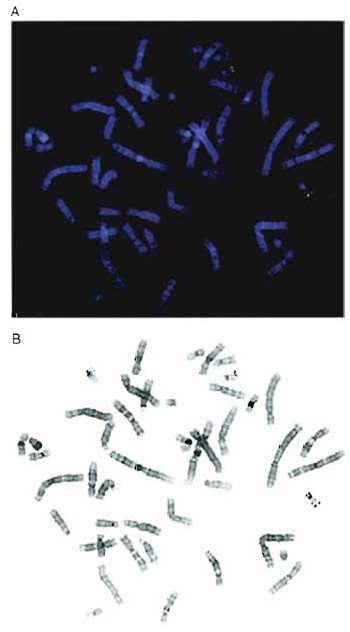 |
| FIGURE 1 (A) Two-color hybridization of two cosmid clones mapping to chromosome 22. One cosmid was biotinylated and detected with avidin-Texas red (red) and the second cosmid was labeled with digoxigenin and visualized using FITC-conjugated antibodies (green). (B) Black-and-white inverted image of the metaphase shown in A. The chromosomes appear essentially G banded, although the 9qh region is dark in this example. |
C. Detection
The 4 × TNFM washes and the immunochemical incubations do not need to be performed at 37-42°C as the results are usually satisfactory at room temperature. However, it is preferable to protect the slides from bright light, and in a busy laboratory the slides can often be tucked away more safely and neatly by leaving them in an incubator or water bath.
If the clones to be mapped all localize to a small region, it may not always be possible to use a doubleprobe protocol as interpretation can be complicated by poor-quality probe. When performing dual-color analysis, it is easier to use fluorochromes that are spectrally well separated. Cy3 is a very stable, bright orange fluorochrome, but it can be difficult to use with FITC, as the Cy3 signals are often visible through FITC filters. This is a particular problem when imaging with black-and-white CCD cameras as strong Cy3 signals can be confused with FITC signals. Texas red is preferred for combinations with FITC (Fig. 1A).
The DAPI staining reveals a clear banding pattern, which permits chromosome identification, provided the slides are not overdenatured. If a digital image of the DAPI-stained metaphase is converted into a blackand- white inverted image, the chromosomes appear similar to a conventional G-banded metaphase (Fig. 1B).
V. PITFALLS
- If no signal is obtained, one possibility may be
insufficient DNA. It is not unusual for spectrophotometric
determinations of DNA concentration to be as
much as 5-10 times higher than the real figure. Alternatively,
the original DNA may have deteriorated.
These problems are monitored easily by running
approximately 100-200ng of the original DNA next to
the 2-µl aliquot of the precipitated nick-translated
product in a standard 1% agarose gel. This will also
reveal whether bacterial clones retain the expected size
insert. When using labeled whole yeast DNA, it may
be necessary to increase the amount of probe used per
hybridization to ensure that there is sufficient YAC probe present. Usually 80-100 ng of whole yeast probe
will ensure clear YAC signals. Using additional
amounts of probe may also assist the mapping of
smaller insert clones.
Another major problem is poor hybridization efficiency because of poor-quality slide preparation. Probe accessibility is critical. Good-quality metaphase preparations, with well-spread chromosomes and a minimum of residual cytoplasm, are far more useful than any amount of protease pretreatment. RNase A digestion is not normally necessary at all.
It is always possible that the clone you are trying to map will never hybridize to your metaphase spreads. For example, human monochromosomal libraries made from material flow sorted from somatic cell hybrids may contain a small proportion of nonhuman clones. Try hybridizing a larger amount of probe (100-200 ng) without any Cot-1 DNA and no preannealing at 37°C. The abundant Alu short, interspersed repeat sequences are distributed throughout the human genome and should be present in most genomic clones. In the absence of preannealing, Alu signals should be distributed all over the chromosomes, concentrated in the G-light chromosome R bands.
It also appears that about 25-30% of cDNA clones cannot be localized by FISH (Korenberg et al., 1995). This may be due to the relatively short genomic extent of the target gene. Mapping cDNAs is a much more demanding process and every step must be rigorously optimized. Protease digestion around the chromosomal DNA may be useful here, but it must be carefully controlled to avoid loss of chromosome morphology (Fan et al., 1990). More sensitive detection systems, such as the use of tyramides, may also be useful (Raap et al., 1995). - If the cloned DNA does not cut well after nick
translation, the large probe fragments will result in
excessive background hybridization, which is seen as
large, bright spots of signal all over the chromosomes
and nuclei. Large spots of signal that appear to float
above the spreads are nearly always indicative of
excessive probe fragment length. Because DNase I
digestion is affected by contaminants in the DNA, it
may be necessary to repurify the sample before repeating
nick translation. DNA that digests with restriction
enzymes should be satisfactory. Try increasing the
duration of the next nick translation reaction by at least
10 min. Check the concentration of your DNA also; the
labeling reaction may have contained far too much
DNA.
If the probe length is in the acceptable size range, it may be that one of the immunochemicals has deteriorated. These should always be kept in small aliquots and handled as aseptically as possible. Depending on the manufacturers' recommendations, it should be possible to store most reagents at -20°C with a single working aliquot stored at 4°C. Avoid repeated freeze-thawing and, for this reason, do not store reagents in frost-free freezers. Using immunochemicals at too high a concentration can result in generalized nonspecific background over the slide. Similarly, precipitation of immunochemical complexes will occur if the slides are allowed to dry out during the detection procedure.
Certain clones may require increased amounts of Cot-1 DNA to suppress apparently nonspecific hybridization to other chromosomal regions. Another potential source of nonspecific background may be residual material in the fixed metaphase suspensions. This may be reduced by washing the cells in several changes of methanol/acetic acid fixative before preparing more slides. We routinely postfix slides in a Coplin jar of fixative, air dry, and then dehydrate through a fresh ethanol series before a final fixation in acetone.
Acknowledgment
My thanks to Dr. Nigel Carter for useful discussions over the manuscript.
References
BAC Resource Consortium (Cheung et al.) (2001). Integration of cyeogeneeic landmarks into the draft sequence of the human genome. Nature 409, 953-958.
Bailey, J. A., Gu, Z., Clark, R. A., Reinere, K., Samonte, R. V., Schwartz, S., Adams, M. D., Myers, E., W., Li, E W., and Eichler, E. E. (2002). Recent segmental duplications in the Human Genome. Science 297, 1003-1007.
Fan, Y.-S., Davis, L. M., and Shows, T. B. (1990). Mapping small DNA sequences by fluorescence in situ hybridization directly on banded metaphase chromosomes. Proc. Natl. Acad. Sci. USA 87, 6223-6227.
Inazawa, J., Ariyama, T., Tokino, T., Tanigami, A., Nakamura, Y., and Abe, T. (1994). High resolution ordering of DNA markers by multi-color fluorescent in situ hybridization of prophase chromosomes. Cytogenet. Cell Genet. 65, 130-135.
Korenberg, J. R., Chen, X.-N., Adams, M. D., and Veneer, J. C. (1995). Towards a cDNA map of the Human Genome. Genomics 29, 364-370.
Lawrence, J. B., Singer, R. H., and McNeil, J. A. (1990). Ineerphase and metaphase resolution of different distances within the human dystrophin gene. Science 249, 928-932.
Raap, A. K., Florijn, R. J., Blonden, L. A. J., Wiegane, J., Vaandrager, J.-W., Vrolijk, H., den Dunnen, J., Tanke, H. J., and van Ommen, G.-J. (1996). Fiber FISH as a DNA mapping tool. Methods 9, 67-73.
Raap, A. K., van de Corput, M. E, Vervenne, R. A., van Gijlswijk, R. P., Tanke, H. J., and Wiegant, J. (1995). Ultra-sensitive FISH using peroxidase-mediated deposition of biotin- or fluorochrome tyramides. Hum. Mol. Genet. 4, 529-534.
Selleri, L., Eubanks, J. H., Giovannini, M., Hermanson, G., Romo, A., Djabali, M., Maurer, S., McElligott, D. L., Smith, M. W., and Evans, G. A. (1992). Detection and characterization of "chimeric" yeast artificial chromosome clones by fluorescent in situ suppression hybridization. Genomics 14, 536-541.
Trask, B. J., Massa, H., Kenwrick, S., and Gitschier, J. (1991). Mapping of human chromosome Xq28 by two-color fluorescence in situ hybridization of DNA sequences to interphase cell nuclei. Am. J. Hum. Genet. 48, 1-15.
Suggested Reading
Carter, N. R (1996). Fluorescence in situ hybridization--state of the art. Bioimaging 4, 41-51.
Lawrence, J. B. (1990). A fluorescence in situ hybridization approach for gene mapping and the study of nuclear organization. In "Genome Analysis (K. E. Davies and S. Tilghman, eds.), Vol. 1, pp. 1-38. Cold Spring Harbor Laboratory Press, Cold Spring Harden, NY.
Lichter, P., and Cremer, T. (1992). Chromosome analysis by nonisotopic in situ hybridization. In "Human Cytogenetics: A Practical Approach" (-D. E. Rooney and B. H. Czepulkowski, eds.), Vol. I., pp.157-192. Oxford Univ. Press, Oxford.
Support our developers




