Northern Blotting
A. Formaldehyde agarose gel electrophoresisFormaldehyde vapors are toxic and casting this gel should be performed in a fume hood. The gel tank must be covered when in use.
Preparation of Equipment and Reagents
Soak the gel tanks, combs etc. in 0.2 M NaOH for 15 minutes to destroy any contaminatig RNases before rinsing in distilled water. It is not necessary to use DEPC-treated water.
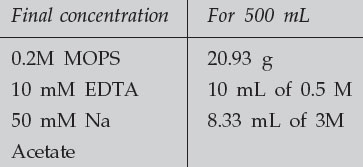
Make up 10 X Northern Running Buffer (containing MOPS)
 |
|---|
The buffer needs to be brought to a pH of 7.0. For 500 mL of 10 X MOPS, this means add approximately 2 g of NaOH. Make up to 500 mL in DEPC-treated water to DEPC-treat solutions, add 0.1% DEPC to the solution in a bottle which can be autoclaved. Mix the solution well and allow it to stand, with the cap tightly closed, for at least 30 minutes. Then loosen the cap and autoclave. This must be done in a fume hood, as DEPC is very toxic. Note also that DEPC is inactivated by water. Allow the DEPC stock solution bottle to warm to RT before opening to avoid condensation.
Autoclaving of the MOPS buffer is not necessary and turns the solution yellow. Store at 4°;C.
Cast the gel
Cast a 14-cm, 0.7–1.0% agarose gel, which requires at least 100 mL of agarose solution.
Ethidium bromide is not added to the gel, but rather to the sample buffer. A thin, low-percentage agarose (0.6%–0.7%) gel is critical for good transfer of large MW RNAs (>4–5kb).
 |
|---|
Dissolve the agarose in the microwave, let the solution cool to less than 60.25°;C, then add the formaldehyde and cast the gel.
Prepare RNA Samples for Loading
RNA is stored at –80.25°;C as an ethanol precipitate. Determine the amount of RNA to be loaded in each well (e.g., 2 mg of poly A(+) RNA). Based on your RNA yield estimations, precipitate the appropriate amount of RNA ethanol solution for at least 15 minutes at 13000 g at 4°;C. Remove all supernatant and resuspend each sample in 12 mL of sample buffer.
RNA sample buffer (prepared fresh from frozen stocks): For 500 mL:
- 50 µL 10 X running buffer
- 250 µL deionized formamide
- 90 µL formaldehyde
- 108 µL water (DEPC-treated)
- 2 µL ethidium bromide (stock concentration, 10 mg/mL)
Running the Gel
Fill the tank with 1X MOPS (prepare 1X with DDW). Early protocols added formaldehyde to the running buffer, but this is unnecessary. Run the gel at between 100 to 200 V for several hours, until the xylene cyanol dye front has migrated 3 to 4 cm into the gel and the bromophenol blue is about two-thirds down the gel. Circulate the buffer from end to end every half an hour, especially if running the gel at 200 V. When the RNA has run an appropriate distance, photograph the gel, including a ruler aligned with the wells. Presoak the gel in 20 X SSC while setting up the transfer (about 15 minutes).
B. Northern Transfer
Setting up Transfer
The physical set-up for Northern transfer is identical to that for Southern transfer, except that 20X SSC is used as the transfer buffer, (Note: RNA is hydrolyzed in strongly alkaline solutions within seconds!) and the membrane we use is Gene Screen Plus. Prewet membrane in DDW and then in 20 X SSC.
Post-transfer Handling of the Membrane
After overnight transfer, the position and orientation of the wells is marked on the membrane (#1 etc.) and it is rinsed gently in 2X SSC for 5 minutes before being air dried in the fume hood. The membrane is baked at 80°;C for 2 hours and is then ready for prehybridization and hybridization.
Method
- Run Northern in the usual way, transfer to Genescreen plus in 20 X SSC, and bake in the oven at 80°;C for 2 hours.
- Hybridization buffer is as follows: 50% Formamide, 3 X SSC, 10X
Denhardt’s, 10 µM phosphate buffer, pH 8.0, 2 µM EDTA, 0.1% SDS,
200 mg/mL herring sperm DNA.
- 800 U/µL preservative-free sodium heparin.
- Prehybridize membrane for 4–6 hours at 60°;C.
- Hybridize in fresh buffer for 18–24 hours at 65°;C–20 ng of riboprobe to the bag in a small volume of buffer (e.g., 3–5 µL). Riboprobes should be prepared exactly as described for hybridization histochemistry. This can include hydrolysis, but this may or may not make a difference to the degree of background.
- Wash at high stringency, i.e., 0.1 X SSC, 0.1% SDS at 65°;C. Be careful to wash all formamide-containing hybridization buffer off at low stringency first, i.e., use a 2 X SSC wash initially, then increase stringency progressively. A good signal can be obtained with little background using this washing regimen, but is lost when washed at 75°;C.
- Expose against x-ray film using an intensifying screen at –70°;C for 24 hours or longer. A good signal can be obtained after 36 hours, equal to that obtained with Northerns using cDNA probes after 96 hours of exposure.
Support our developers




